The basic approach is to use Beer’s law to measure the concentration of a protein solution:
![]()
A280 is the absorbance of a protein solution at 280 nm.
ε is the molar extinction coefficient (in 1/(M*cm)). This value describes how much 280 nm light a one molar protein solution will absorb over a 1 cm cell.
l is the pathlength in cm. Our spectrophotometer has a pathlength of 1 cm; the nanocuvette has a pathlength of 0.1 cm.
c is the protein concentration in molar units.
Using the Eppendorf Spectrophotometer
- Step 1
- Step 2
- … etc.
Scattering correction
The apparent absorbance at 280 nm has contributions from the absorption of aromatic residues (what we want), as well as from light scattered by aggregate in solution (what we don’t want). To measure the scattering contribution, we measure the absorbance at 320 nm and 340 nm — where aromatic absorption is minimal, meaning that any signal we observe arises from scatter — and then extrapolate back to find the scattering contribution at 280 nm. Scatter increases as λ^(-4), so the extrapolation is done on a log/log scale:

An excel spreadsheet implementing this correction is here. The paper describing this procedure is here.
This should also be viewed as a quality check: assuming the cuvette is clean, scatter arises from aggregate in solution, which will likely interfere with your experiments. If scattering >0.01 is observed, centrifuge the sample for 10 min @ 13,000 rpm and then pipette the supernatant into a fresh tube. (Centrifuge in the temperature controlled centrifuge, as samples can heat up significantly in the standard microcentrifuges). Re-measure the scattering contribution.
Extinction Coefficient
This can be estimated from the sequence of the protein by assuming an additive contribution from each Trp, Tyr, Phe, and Cys residue in the protein. Protparam is useful server for doing this calculation.
Unfortunately, the additive assumption does not always hold, particularly for proteins that do not contain tryptophan. We therefore must measure a “corrected” extinction coefficient. The additive assumption holds for a protein fully denatured in 6 M GdnHCl. We can use this fact, plus the fact that absorbance and extinction coefficient are directly proportional, to determine the extinction coefficient of the protein in 0 M GdnHCl.
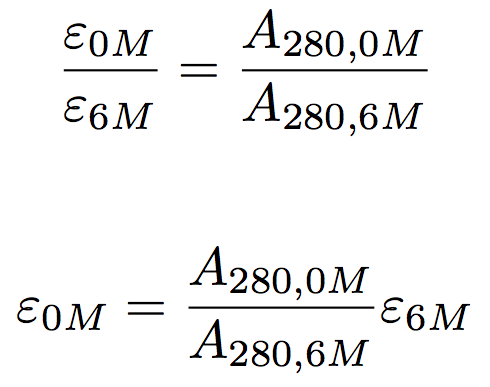 We therefore need to measure the absorbance of a fixed amount or protein in the absence and presence of 6 M GdnHCl to then correct the theoretical extinction coefficient.
We therefore need to measure the absorbance of a fixed amount or protein in the absence and presence of 6 M GdnHCl to then correct the theoretical extinction coefficient.
This can be done, as follows.
- Obtain ~100 uL of pure protein stock with an A280 > 0.5 (preferably closer to 1.0) and no detectable scatter.
- Make buffer stocks at 0 M and 6 M GdnHCl.
- Add 20 uL protein to 180 uL stock. Mix thoroughly. This has to be accurate, so do it with Hamilton syringes. Wait 10 minutes to make sure that the protein in the 6 M GdnHCl solution is fully denatured.
- Blank the spectrophotometer with 0 M GdnHCl and measure A280 of 0 M GdnHCl protein.
- Blank with 6 M GdnHCl buffer. The high GdnHCl concentration will cause refractive index “swirls”, so you should rinse the cuvette 3-5 times with the GdnHCl buffer before blanking and doing the protein measurement.
- Use the equation above to estimate the extinction coefficient in 0 M GdnHCl given the theoretical extinction coefficient and absorbance measured in 0 and 6 M GdnHCl.
- Repeat full protocol 2 more times. If your technique is good, the standard error should be tiny relative to the extinction coefficient.
This protocol only has to be done once for a given protein.